Blood / Hematology
The latest blood and hematology research from prestigious universities and journals throughout the world.
Blood consists of a liquid called plasma, red blood cells, white blood cells and platelets. Red bloods cells deliver oxygen from your lungs to your tissues and organs, white blood cells fight infection as part of your body's defense system, and platelets help blood clot when you experience a cut or wound. Blood is a constantly circulating fluid providing the body with nutrition, oxygen, and waste removal. Your blood group depends on which antigens occur on the surface of your red blood cells.
What is Hemophilia? What is Haemophilia?

Many members of royalty in Europe inherited their hemophilia
from Queen Victoria".
This is a file from Wikimedia Commons
Hemophilia is a group of inherited blood disorders in which the blood does not clot properly. Hemophilia is the standard international spelling, also known as haemophilia in the UK, other translations include: hémophilie, hemofilie, hemofili, hemofilia, hämophilie, emofilia. We will use the standard international spelling for the purpose of this section.
Bleeding disorders are due to defects in the blood vessels, the coagulation mechanism, or the blood platelets. An affected individual may bleed spontaneously or for longer than a healthy person after injury or surgery.
The blood coagulation mechanism is a process which transforms the blood from a liquid into a solid, and involves several different clotting factors. The mechanism generates fibrin when it is activated, which together with the platelet plug, stops the bleeding.
When coagulation factors are missing or deficient the blood does not clot properly and bleeding continues.
Patients with Hemophilia A or B have a genetic defect which results in a deficiency in one of the blood clotting factors.
Queen Victoria was a carrier and passed the mutation to her son Leopold, and through several of her daughters to members of the royal families of Spain, Russia, and Germany.
Tsarevich Alexei Nikolaevich, son of Nicholas II (Russia) suffered from hemophilia and was a descendant of Queen Victoria - Rasputin was successful in treating his hemophilia, it was claimed.
Types of Hemophilia / Haemophilia
Hemophilia A and Hemophilia B
There are two main types of hemophilia - Hemophilia A (due to factor VIII deficiency) and Hemophilia B (due to factor IX deficiency). They are clinically almost identical and are associated with spontaneous bleeding into joints and muscles and internal or external bleeding after injury or surgery.
After repeated bleeding episodes permanent damage may be caused to the joints and muscles that have been affected, particularly the ankles, knees and elbows.
Approximately 1 in 5,000 males is born with Hemophilia A, and 1 in 30,000 males is born with Hemophilia B. Hemophilia affects people of all races and ethnic origins globally. The conditions are both X-linked and virtually all sufferers of hemophilia are males. Female carriers may also bleed abnormally, because some have low levels of the relevant clotting factor.
People with hemophilia have a genetic mutation in the affected gene on the X chromosome, which results in reduced production of Factor VIII or IX and creates a bleeding tendency, because coagulation takes much longer than normal, thus making the clot weak and unstable
Approximately one third of patients with hemophilia have no family history of the disease, either because of new genetic mutations, or because previous affected generations either had daughters (who were carriers) or sons who died in early childhood from hemophilia or any other cause or who were not affected.
Acquired hemophilia
This is very rare. The patient develops the condition during his/her lifetime and it does not have a genetic or heritable cause. It occurs when the body forms antibodies that attack one or more blood clotting factors, (usually factor VIII), thus preventing the blood clotting mechanism from working properly. Patients may be male or female and the pattern of bleeding is rather different from that of classical hemophilia, the joints being rarely affected. The disorder is particularly associated with old age and occasionally complicates pregnancy.
What Causes Hemophilia / Haemophilia?
People with hemophilia are born with it. It is caused by a fault in one of the genes that determine how the body makes blood clotting factor VIII or IX. These genes are located on the X chromosome.
To understand how hemophilia is inherited, it is important to learn about chromosomes.
What are chromosomes?
Chromosomes are blocks of DNA (deoxyribonucleic acid). They contain very detailed and specific instructions that determine:
-
How the cells in a baby's body develop.
-
What features the baby will have, including, for example, hair and eye color.
-
Whether the baby is male or female.
In humans there are 23 pairs of chromosomes, including the sex chromosome pair. There are two types of sex chromosome:
-
The X chromosome
-
The Y chromosome
All humans have a pair of sex chromosomes:
-
Males have an X + Y pair
-
Females have an X + X pair
NB Females do not have any Y chromosomes.
What chromosomes do we inherit from our parents?
-
A Male inherits his
-
X chromosome from his mother
-
Y chromosome from his father
-
-
A Female inherits
-
One X chromosome from her mother
-
One X chromosome from her father
She does not inherit both X chromosomes from her mother. She has no Y chromosomes.
-
How can we calculate the risk of hemophilia in offspring?
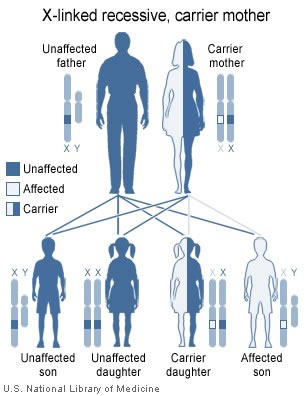
(Before reading on, remember that the faulty gene is never on the Y chromosome. If it is present, it will be on the X chromosome.)
-
Female (X + Xfaulty) is a carrier, but does not have hemophilia. The “good” X chromosome allows the production of enough clotting factor to prevent serious bleeding problems.
-
Male (Y + Xfaulty) will develop hemophilia and can pass it on.
If the father has hemophilia and the mother has no faulty gene (is not a carrier):
Father (Y + Xfaulty). Mother (X + X).
-
There is no risk of inherited hemophilia in their sons because boys will inherit their X chromosome from the mother, not the father (they inherit the father's Y chromosome only, which does not have the faulty gene).
-
All the daughters will be carriers but will not develop hemophilia although they will inherit the father's X chromosome, which has the faulty gene. However, their maternal X chromosome, which does not have the faulty gene, usually allows the production of enough clotting factor to prevent serious bleeding problems.
If the father does not have hemophilia and the mother has a faulty gene:
Father (Y + X). Mother (X + Xfaulty).
-
There is a 50% chance that sons will develop hemophilia because:
-
There is a 50% risk that a son will inherit his mother's Xfaulty chromosome, plus his father's Y chromosome - he will have hemophilia.
-
There is a 50% chance he will inherit his mother's "good" X chromosome, plus his father's Y chromosome - he will not have hemophilia.
-
-
There is a 50% chance that daughters will be carriers, (but no chance of developing hemophilia), because:
-
There is a 50% chance she will inherit her mother's Xfaulty chromosome, making her a carrier.
-
-
There is a 50% chance she will inherit her mother's "good" X chromosome, which would mean she would not be a carrier.
Approximately one third of patients with hemophilia have no family history of the disease, either because of new genetic mutations, or because previous affected generations either had daughters (who were carriers) or sons who died in early childhood from hemophilia or any other cause or who were not affected.
What is Coagulation? How does our blood clot?
Coagulation is a complex process by which the blood forms clots to block and then heal a lesion/wound/cut and stop the bleeding. It is a crucial part of hemostasis - stopping blood loss from damaged blood vessels. In hemostasis a damaged blood vessel wall is plugged by a platelet and a fibrin-containing clot to stop the bleeding, so that the damage can be repaired.
Coagulation involves a cellular (platelet) and protein (coagulation factor) component.
When the lining of a blood vessel (endothelium) is damaged, platelets immediately form a plug at the site of the injury, while at the same time proteins in the blood plasma respond in a complex chemical reaction, rather like a waterfall, to form fibrin strands which reinforce the platelet plug.
Primary hemostasis - when the platelets gather at the site of an injury to plug (block) it.
Secondary hemostasis - proteins (coagulation factors) act in a series of chemical reactions to strengthen the plug and allow healing to begin.
What is a platelet?
A platelet is a disc-shaped element in the blood that is involved in blood clotting. They aggregate (clump together) during normal blood clotting. They are classed as blood cells, but are in fact fragments of large bone marrow cells called megakaryocytes.
What is fibrin?
Fibrin is an insoluble protein involved in blood clotting. Fibrin is deposited around the wound in a form of mesh to strengthen the platelet plug. The whole thing dries and hardens (coagulates) so that the bleeding stops and the wound then heals. Fibrin is developed in the blood from a soluble protein, fibrinogen.
When platelets come into contact with damaged tissue thrombin is formed as a result of a series of chemical processes (coagulation cascade) that culminate in the formation of fibrin from fibrinogen.
Coagulation factors (clotting factors)
Coagulation factors are proteins, mostly manufactured by the liver. They were originally numbered in the order of their discovery, traditionally using Roman numerals from I-XIII. Some of the numbers such as III and VI are not used any more and in recent years, many proteins that affect blood clotting have been discovered but have been given a name rather than a number. When a blood vessel wall is damaged, or any kind of wound occurs, a complex set of chemical reactions involving these coagulation factors (and acting rather like a waterfall) takes place.
The final step of the cascade of chemical reactions is to convert fibrinogen - Factor I - into fibrin, forming a mesh which clumps platelets and blood cells into a solid clot, plugging the hole and stopping the bleeding. Patients with Hemophilia A have deficient levels of Factor VIII, while patients with Hemophilia B have deficient levels of Factor IX.
Hemophilia Symptoms and Diagnosis
What are the symptoms of hemophilia?
Hemophilia symptoms vary, depending on the degree of blood clotting factor (coagulation factor) deficiency and they also depend on the nature of any injury.
Three levels of hemophilia are recognized, according to the level of clotting factor amounts in the blood. These are often expressed as percentages of normal:
-
Above 5% - mild hemophilia
-
1% to 5% - moderate hemophilia
-
Less than 1% - severe hemophilia
Mild hemophilia
People with inherited mild hemophilia may not have any symptoms until an event occurs which wounds the skin or tissue, such as a dental procedure or surgery, and results in prolonged bleeding. In societies where male circumcision is carried out soon after birth, mild hemophilia will be detected earlier. Joint bleeding is uncommon.
Moderate hemophilia
Those with inherited moderate hemophilia will be noticeable early on. The child will bruise easily and may also experience internal bleeding symptoms, especially around the joints, and after a blow or a fall. Bleeding that occurs inside a joint is usually referred to as a joint bleed.
Symptoms of a joint bleed:
-
Tingling sensation in the joint
-
Pain in the joint
-
Irritation in the joint
If left untreated, the patient may eventually experience:
-
More severe pain in the joint
-
Joint stiffness
-
The affected area becomes swollen, tender and hot
Joint bleeds most commonly affect the:
-
Ankles
-
Knees
-
Elbows
...and may less commonly affect the shoulders, hips or other joints.
Any surgical intervention, circumcision, dental procedure or injury will result in prolonged bleeding in a person with hemophilia.
Severe hemophilia
Symptoms are similar to those found in moderate hemophilia, but occur more frequently and are usually more severe.
A child with severe hemophilia will often bleed for no apparent reason, often referred to as spontaneous bleeding. Most commonly, in early childhood from about 18 months of age, the nose or mouth start to bleed or apparently spontaneous bruises appear, particularly on the legs. Parents are sometimes suspected of causing non-accidental injury (deliberate harm) to their children.
Symptoms of hemophilia type bleeding may include:
-
Several large or deep bruises
-
Joint pain or swelling
-
Unexplained bleeding or bruising
-
Blood in feces (stools)
-
Blood in urine
-
Unexplained nosebleeds
-
Unexplained gum bleeding
-
Tightness in the joints
Intracranial hemorrhage (bleeding inside the skull)
About 1 in every 30 patients with hemophilia will have intracranial hemorrhage at least once during their lives. This should be treated as a medical emergency. Spontaneous intracranial hemorrhage is rare and in many cases bleeding inside the skull will be the result of a blow to the head.
Symptoms of intracranial hemorrhage include:
-
A bad headache
-
Vomiting
-
Confusion
-
Fitting (Convulsion)
-
Loss of balance
-
Slurred speech, or other speaking difficulties
-
Stiff neck
-
Vision problems
-
Loss of coordination
-
Some of the facial muscles do not work (sometimes all of them)
How is hemophilia diagnosed?
Prenatal testing - if a pregnant woman has a history of hemophilia, a hemophilia gene test can be done during pregnancy. A sample of placenta is removed from the uterus and tested. This test is known as a CVS (chorionic villus sampling) test.
Blood test - if a doctor suspects a child may have hemophilia a blood test can determine whether the patient has hemophilia A or B, and how severe it is. Blood tests can be performed from the time of birth onwards.
Treatment for Hemophilia / Haemophilia

Up to a few decades ago a considerable proportion of patients with hemophilia died prematurely because of hemophilia. Tragically, many deaths were the result of childhood injury or surgery. Over the last forty years treatment has advanced so much that the vast majority of patients today are expected to live long and active lives.
The main breakthrough in treatment occurred when coagulation factor deficiencies linked to hemophilia could be identified and then replaced, using products derived from human blood.
In the past patients used to receive whole blood or plasma infusions to control episodes of bleeding. Even though this helped, levels of clotting factors, especially factors VIII and IX, never reached the levels required for really effective blood coagulation, nor could these levels be sustained - in other words, serious bleeding was only partly treated.
Cryoprecipitate, made through the cold precipitation of frozen plasma from1965 onwards, was the first really effective treatment for hemophilia A. Freeze-dried concentrates made from human plasma containing the right levels of Factors VIII and IX became available in the late 1960s and early 1970s. Being able to keep the treatment at home and use it as required meant that patients could travel, leave the home, go to work, and enjoy a level of independence. However, a large number of patients subsequently became infected with blood-borne pathogens, such as hepatitis B, hepatitis C and HIV.
From the mid 1980s rigorous donor selection and viral inactivation procedures reduced the risk of blood-borne viral transmission to nearly zero. During the 1990s it became possible to prepare synthetic (recombinant) factors, using specially prepared mammalian cells and these recombinant concentrates are now widely used.
Hemophilia treatment will mainly depend on its severity and for patients with Hemophilia A or B involves clotting factor replacement therapy. There are two approaches:
-
On demand - giving treatment to stop prolonged bleeding when it occurs. This is more common in the management of patients with mild hemophilia.
-
Preventative treatment (prophylaxis) - medication to prevent bleeding episodes, and subsequent complications, such as joint and/or muscle damage. More commonly used for patients with moderate or severe hemophilia.
Clotting factor concentrates
Clotting factor concentrates can be made in two different ways:
-
Plasma-derived clotting factors - prepared from the plasma of donated human blood.
-
Recombinant clotting factors - the first generation of recombinant products use animal products in the culture medium and had human albumin (a human blood product) added as a stabiliser. Second generation products use animal-derived materials in the culture medium but do not have added albumin and instead use sucrose or other non-human derived material as a stabiliser. Third generation clotting factors have no albumin present at any stage of their preparation. Mouse monoclonal antibodies have been routinely used in the purification of coagulation factors for many years but a recently licensed recombinant factor VIII employs a synthetic ligand for this step. This has resulted in the production of the first factor VIII concentrate to be free of all exogenous human and animal protein, a goal which was reached for hemophilia B when the first recombinant factor IX was licensed in 1997.
Desmopressin (DDAVP)(for mild hemophilia A)
This medication is a synthetic hormone which encourages the body to produce more of its own Factor VIII. It is unsuitable for patients with hemophilia B and those with severe hemophilia A. In patients with milder forms of hemophilia A, factor VIII replacement therapy may be necessary, especially for severe bleeds, or after serious injury or major surgery.
RICE (Rest, Ice, Compression, Elevation)
RICE is a treatment many health care professionals recommend for joint bleeds. It also reduces swelling and tissue damage when used together with clotting factor concentrates.
Administering clotting factor concentrates
The medication is injected into a vein - generally in the back of the hand or at the crook of the elbow. Initial treatments are usually administered by a doctor or nurse at a hospital or clinic. Most adults can learn how to do this themselves, which means they can stop bleeding rapidly and effectively wherever they are.
If the patient is a child the parents or caregivers (UK/Ireland/Australia: carers) can learn how to administer treatment. The majority of very young patients can receive most of their treatment at home.
If a patient is finding it hard to access a suitable vein, or if intensive treatment is required, a port-a-cath, or an external catheter called a Broviac or Hickman line can be placed surgically into a vein, allowing factor replacement therapies to be given, and blood to be drawn easily for routine emergency tests. The use of such catheters can be complicated by infection and blockage and they have to be used with great care.
Treating bleeds
Bleeding episodes (bleeds) are an inevitable complication for patients with hemophilia A and B, even for patients with mild forms. As the underlying problem is one of prolonged bleeding, rather than rapid bleeding, they often appear not to be medical emergencies.
If a person with hemophilia experiences any of the following he should seek immediate skilled medical help:
-
There is an injury to the neck, mouth, tongue, face or eye.
-
There is a severe blow to the head.
-
Bleeding is heavy or persistent.
-
There is severe pain or swelling in any part of the body.
-
An open wound requires stitching.
Most other bleeds, such as joint/muscle bleeds, small injuries and cuts that do not require stitches, and nosebleeds are generally treated at home, but patients should always seek the advice of a healthcare professional when in doubt. Any treatment will be more effective if it is started early.
Storing treatment
Factor concentrates should usually be stored in a refrigerator but are stable at room temperature for quite long periods. They should not be frozen as this may damage the vials or syringes. Some may be taken out for travel but should ideally be kept in a cool bag. Read instructions on product storage. If you are unsure, check with a health care professional or qualified pharmacist.
Inhibitors
Approximately 30% of people with severe hemophilia A develop antibodies to transfused factor VIII, usually shortly after their first few treatments. These antibodies (also called inhibitors) prevent the factor VIIII treatment working properly. It is often the case that, after a while, the inhibitors disappear and only about 10% or less of people with severe hemophilia A will suffer from long term inhibitors. In recent years it has become possible to prevent inhibitors becoming persistent through immune tolerance induction therapy. Where inhibitors do not respond to this approach alternative treatments are available.
Inhibitors rarely develop in mild hemophilia A or in hemophilia B of any severity.
This Hemophilia information section was written by Christian Nordqvist for Medical News Today, and may not be re-produced in any way without the permission of Medical News Today.
Additional materials provided by Wyeth.